
Raziskovanja v biologiji si danes ne moremo več predstavljati brez računalnikov. Potrebujemo jih ne glede na to, na kateri stopnji kompleksnosti raziskujemo življenjske procese. Pomagajo nam pridobivati podatke, jih urejati in analizirati. Zaradi hitrega razvoja tehnologije in raziskovalnih metod v zadnjih desetih letih smo v biokemiji, molekularni in celični biologiji sposobni podatke pridobivati z neverjetno hitrostjo. Kot pomoč pri zajemanju, urejanju in analizi raznovrstnih bioloških podatkov se je razvila nova znanstvena disciplina – bioinformatika.
Biolog v obdobju genomike
Predstavljajte si, da bi se Ljubljana povečala za dvakrat vsakega pol leta. Že čez nekaj let bi bila metropolis, podobna Londonu ali New Yorku. Predstavljajte si še, da v tem času ne bi mogli dovolj hitro izdelati dobrih mestnih zemljevidov ali telefonskih imenikov. Verjetno bi bilo kakršnokoli iskanje, npr. najboljšega servisa za vaš avtomobil, prava mora. V podobnem stanju so zadnjih nekaj let nekatere biološke znanosti, kjer smo priča neznanskemu razvoju. Največji napredek so doživele biokemija, molekularna biologija in genetika. Ker raziskovalce v teh znanostih zanima predvsem delovanje celice na molekularnem nivoju, ima narava večine bioloških podatkov opraviti predvsem z lastnostmi bioloških makromolekul. To predstavlja prvi nivo kompleksnosti živega (slika 1).
En od vrhuncev razvoja je bil gotovo projekt človeškega genoma, ki je bil največji in najdražji projekt v zgodovini biologije. Rezultati tega projekta so omogočili vpogled v katalog vseh genov in njihovih produktov, proteinov, ki sodelujejo pri organiziranju in delovanju človeških celic. Genom pa niso zgolj geni. Le nekaj odstotkov človeškega genoma nosi zapis za gene. Za znanstvenike je bil velik zalogaj poiskati te majhne kose v morju treh milijard nukleotidov, ki sestavljajo človeški genom. Pomagajo si z novo znanstveno disciplino, bioinformatiko, ki pomaga iz te nepregledne množice podatkov izluščiti smiselne, biološke informacije. Večinoma se bioinformatiki ukvarjajo z iskanjem in analizo proteinskih in nukleotidnih zaporedij in tridimenzionalnih (3D) struktur proteinov, v zadnjem času pa tudi z analizo novih vrst podatkov, ki se kopičijo pri transkriptomskih (npr. izpisi genskih čipov) ali proteomskih projektih (npr. 2D geli). Sprva se je pojem bioinformatike pojavil v začetku devetdesetih let predvsem v povezavi z objavljanjem in urejanjem publikacij na svetovnem spletu. Današnja definicija bioinformatike pravi, da bioinformatika vključuje matematične, statistične in računalniške metode za reševanje bioloških problemov z uporabo nukleotidnih in proteinskih zaporedij in s temi povezane informacije. V prispevku bom prikazal pomen proteinskih in nukleotidnih zaporedij za razumevanje delovanja proteinov, kako v laboratorijih pridobimo takšne podatke, kako so ti podatki urejeni in shranjeni in kako jih lahko preiskujemo.
Proteini kot izvajalci celičnih procesov
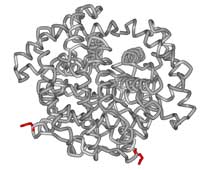
Skoraj vse funkcije žive celice opravljajo proteini. Med drugim so proteini vpleteni v vrsto zapletenih kemijskih reakcij, ki omogočajo pridobivanje energije in osnovnih gradnikov potrebnih za delovanje in razvoj celice. Zato je razumljivo, da je večji del študij biokemije in molekularne biologije usmerjen v razumevanje delovanja proteinov. Proteini so sestavljeni iz osnovnih gradnikov, aminokislin, ki so med seboj povezane s peptidno vezjo. Nastanejo dolge polipeptidne verige, ki se med seboj razlikujejo po velikosti, t.j. po številu aminokislin in aminokislinski sestavi. Za razumevanje funkcije nekega proteina je izredno pomembno poznavanje njegove zgradbe. Navadno govorimo o večih nivojih organiziranosti proteinskih molekul. Če poznamo zaporedje aminokislin, ki ga gradijo govorimo o primarni zgradbi proteina. Navadno te aminokisline napišemo na papir kot zaporedje črk s posebno “abecedo”, enočrkovno kodo, kjer vsaka črka pomeni točno določeno aminokislino (npr. M za metionin, V za valin, W za triptofan itn.) (slika 2). O tridimenzionalni zgradbi (3D) pa govorimo takrat, ko aminokisline v prostoru zavzamejo točno določeno orientacijo. Takrat se lahko zelo približajo aminokisline, ki v primarni zgradbi niso blizu skupaj. Ustvarijo se žepi in površine, ki so odgovorne za delovanje proteina. Ta se z njimi dotika drugih proteinov ali pa v žepih, če gre za encime, potekajo kemijske reakcije. Katere aminokisline gradijo protein in kako so nameščene v prostoru je pomemben podatek, ki biokemiku močno olajša opisovanje lastnosti in funkcije takšnega proteina. Poznavanje strukture molekul je pomembno tudi v medicini. Pri določenih bolezenskih stanjih pride do mutacij genov, kar se odrazi v spremenjeni zgradbi proteinov. S poznavanjem strukture takšnega spremenjenega proteina lahko zato opišemo bolezen na molekularni ravni in morda tudi razložimo opažene simptome. Naj to ponazorim na primeru hemoglobina (slika 2).
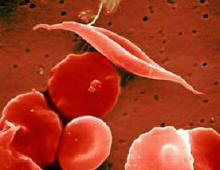
Hemoglobin je molekula, ki po telesu prenaša kisik. Sestavljen je iz štirih podenot. Le-te so razporejene tako, da ima molekula obliko manjše kroglice. V sredi je neproteinski del, molekula hema, kamor je vezan atom železa, ki nase veže kisik. Genov za podenote hemoglobina je v človeškem genomu več. Govorimo kar o globinski družini genov. Nekateri so izraženi v fetalnem in mladostnem obdobju, pri odraslih pa je hemoglobin sestavljen predvsem iz podenot alfa in beta. Pri določenih bolezenskih stanjih so geni za nekatere podenote okvarjeni. Ena takšnih bolezni je srpastocelična anemija, kjer je v beta podenoti šesta aminokislina po vrsti spremenjena zaradi mutacije v genu. Tak hemoglobin so poimenovali hemoglobin S. Mutacija povzroči zamenjavo močno polarne glutaminske kisline v hidrofobno aminokislino valin. Posledica je spremenjena površina molekule hemoglobina, kar privede do kopičenja molekul hemoglobina v podolgovate fibrile, ki popolnoma deformirajo obliko eritrocita (slika 2). Simptome bolezni (spremenjeno obliko eritrocita) so torej lahko razložili s strukturo spremenjenega hemoglobina in agregatov, ki jih tvori. Popolnoma isti pristop se uporablja pri ostalih boleznih, kjer so vpleteni mutirani geni.
Poplava proteinskih in nukleotidnih zaporedij
Za razumevanje življenjskih procesov na molekularni ravni moramo torej poznati primarno in 3D zgradbo proteinov. Zaporedja proteinov lahko določimo na različne načine. če imamo protein izoliran v čisti obliki, lahko določimo primarno zgradbo proteina z uporabo posebne kemijske reakcije, ki postopno odceplja aminokisline od začetka polipeptidne verige proteina proti koncu. Frederick Sanger je bil prvi, ki mu je uspelo po 15 letih mukotrpnega razvijanja postopkov analize in ločevanja majhnih molekul leta 1955 določiti primarno zgradbo hormona insulin. Za razvoj teh postopkov je leta 1958 prejel Nobelovo nagrado za kemijo. Včasih pa proteinov ne moremo pridobiti v dovolj čisti obliki ali v potrebnih količinah. Takrat si pomagamo z določitvijo nukleotidnega zaporedja, saj lahko aminokislinsko zaporedje proteina prevedemo iz nukleotidnega zaporedje gena. Postopke za določevanje nukleotidnega zaporedja DNA so razvili v sedemdesetih letih. Glavni raziskovalec je bil spet Sanger, ki mu je uspelo razviti t.i. Sangerjevo metodo določevanja nukleotidnega zaporedja. Za določitev nekaj sto baznih parov dolgo zaporedje so takrat celi laboratoriji potrebovali nekaj mesecev, nukleotidno zaporedje enega gena pa je bila lahko tema doktorske naloge. Za to reakcijo je Sanger leta 1980 prejel še eno Nobelovo nagrado za kemijo. Sangerjevo metodo uporabljamo skoraj nespremenjeno tudi danes. Razlika je v tem, da je sedaj celoten postopek popolnoma avtomatiziran s pomočjo posebnih robotov in aparatov za določevanje nukleotidnega zaporedja. Najboljši aparati določijo dnevno okoli tisoč zaporedij DNA. Avtomatizacija laboratorijskih postopkov je bila ključna za uspeh pri mnogih genomskih projektih, tudi človeškem. Tako so postavili prave “tovarne” za določevanje nukleotidnih zaporedij. V družbi Celera Genomics so pri določevanju človeškega genoma uporabljali kar 300 takšnih aparatov. Danes so sposobni razvozljati cel bakterijski genom (nekaj milijonov baznih parov) že v enem samem popoldnevu. Posledica takšnega razvoja je, da se število zaporedij, predvsem nukleotidnih, povečuje eksponentno in dosega hitrost okoli milijona in več novih zaporedij na mesec (glej tudi sliko 1).
Podatkovne zbirke v biokemiji in molekularni biologiji
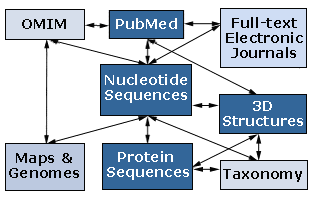
Število proteinskih in nukleotidnih zaporedij se je po začetnih korakih kmalu začelo močno povečevati, zato so jih začeli urejati v posebne podatkovne zbirke. Prva podatkovna zbirka, ki je vsebovala podatke o bioloških makromolekulah je bila “Atlas proteinskih zaporedij in struktur”, ki jo je izdajala v knjižni obliki Margaret Dayhoff v letih 1968-1978. V njej so bila zbrana vsa proteinska zaporedja, uporabljali pa so jo poleg biokemikov in molekularnih biologov tudi raziskovalci, ki so se ukvarjali z molekularno evolucijo. Prva podatkovna zbirka za zaporedja nukleinskih kislin je bila GenBank, ki je bila ustanovljena leta 1982. Prva zaporedja so zaposleni v obeh zbirkah vpisovali iz člankov objavljenih v primarnih znanstvenih publikacijah. Takšne zbirke so razpošiljali raziskovalnim inštitucijam v knjižni obliki ali v obliki magnetnih trakov. Z razvojem poceni osebnih računalnikov in interneta sredi osemdesetih in v začetku devetdesetih let, pa so do podatkov v podatkovnih zbirkah lahko prišli tudi raziskovalci z inštitucij, ki niso imeli neposrednega dostopa do podatkovnih zbirk. Upravljalci zbirk so pripravili uporabniku prijazne dostopne strani in celo iskalna orodja s katerimi lahko po zbirkah iščemo ali brskamo. Podatkovne zbirke danes delimo na bibliografske, primarne, sekundarne in strukturne (tabela 1 in slika 3).
V primarnih podatkovnih zbirkah so shranjena primarna zaporedja proteinov ali zaporedja nukleinskih kislin. Najbolj znana primarna zbirka nukleotidnih zaporedij je že omenjena GenBank, obstajajo pa tudi druge, npr. EMBL v Evropi in DDBJ na Japonskem. Najbolj znana podatkovna zbirka proteinskih zaporedij je SwissProt. Ta podatkovna zbirka vsebuje poleg samega proteinskega zaporedja tudi kopico drugih informacij, npr. bibliografske, kdaj so protein odkrili, nekaj o njegovih lastnostih, kje se izraža ipd, ter povezave na druge podatkovne zbirke (na sliki 4 je prikazan zapis za beto podenoto hemoglobina). Zaradi te dodatne informacije velja SwissProt kot najboljša in zato standardna podatkovna zbirka za proteine.
Sekundarne podatkovne zbirke so iz primarnih izpeljane podatkovne zbirke, ki vključujejo že neko določeno znanje. Vsebujejo npr. samo koščke proteinskih ali nukleotidnih zaporedij DNA. Takšni delčki so navadno zelo pomembni za delovanje proteinov. Npr. katalitska mesta encimov sestavljajo točno določene aminokisline. Te so navadno popolnoma ohranjene pri vseh encimih, ki katalizirajo isto reakcijo. Če torej najdemo takšen košček proteinskega zaporedja v nekem neznanem proteinu lahko sklepamo na to, da je ta protein encim in da katalizira točno določeno reakcijo. Takšen košček je torej nekakšen prstni odtis proteina. Pri proteinih jih imenujemo profili, motivi ali bloki. V DNA zaporedjih so takšni koščki lahko npr. vezavna mesta za proteine, ki sodelujejo pri procesih prepisovanja ali podvajanja DNA. Strukturne podatkovne zbirke hranijo podatke o tridimenzionalni strukturi proteinov ali nukleinskih kislin.
Strežniki za preiskovanje podatkovnih zbirk
Leta 1988 so v ZDA zaradi poplave različne biološke informacije ustanovili Nacionalni Inštitut za Biolotehnološko Informacijo (NCBI). Poslanstvo NCBI-ja je urejanje podatkovnih zbirk z biološko vsebino, razvijanje novih programov za analizo podatkov in tudi izobraževanje s področja bioinformatike. Na NCBI so prestavili GenBank, dosegli pa so tudi to, da je MEDLINE postala primarna bibliografska podatkovna zbirka za področje molekularne biologije. NCBI je dosegel troje: povezal je PubMed, javno dostopen del MEDLINE-a, z ostalimi primarnimi podatkovnimi zbirkami, PubMed so povezali s stranmi založnikov tako, da lahko nekatere članke prebiramo v polnem tekstu in MEDLINE je preko PubMeda postal prosto dostopen preko interneta. V slabih petnajstih letih so ustanovili in uredili tudi ostale podatkovne zbirke in jih povezali v omrežje predstavljeno na sliki 5. Zapisi v posameznih podatkovnih zbirkah so navzkrižno povezani z ostalimi zbirkami, preiskovati pa se jih da preko posebej pripravljenega strežnika, Entrez-a. Zaradi povezovanja različnih podatkovnih zbirk postaja Entrez eden najbolj popularnih strežnikov za iskanje informacije v morju shranjenih podatkov. Takšno povezovanje primarnih podatkovnih zbirk (proteinskih in DNA zaporedij) z ostalimi podatkovnimi zbirkami lahko najdemo tudi v Evropi (SRS – Sequence Retrieval System) in na Japonskem (DBGET na GenomeNet ).
Iskanje zapisov v podatkovnih zbirkah
Informacijo shranjeno v podatkovnih zbirkah lahko preiskujemo na različne načine. Preiskujemo lahko s pristopnimi številkami, ključnimi besedami ali pa, najpogosteje, kar s samim zaporedjem. Navadno je postopek tak, da poskušamo z neznanim proteinskim ali nukleotidnim zaporedjem, ki smo ga določili v laboratoriju, preiskati podatkovne zbirke in na ta način preveriti ali že obstaja kakšen podoben protein in kakšne so njegove lastnosti. Postopek iskanja se začne na vstopnih straneh podatkovnih zbirk z vpisom našega zaporedja, ki ga imenujemo sonda. Računalnik vsak zapis v podatkovni zbirki primerja z našim zaporedjem. V nekaj sekundah se nam na zaslonu računalnika izpišejo rezultati iskanja v oblikiporavnav. Poravnava je poseben zapis dveh ali več zaporedij. Zaporedja izpišemo eno pod drugo tako, da iste ali podobne aminokisline pridejo na enaka mesta. Na sliki 6 je prikazana primerjava zaporedja b podenote hemoglobina in mioglobina, ki je hemoglobinom soroden protein.
Vidimo, da so na nekaterih mestih identične ali po kemijskih lastnostih zelo podobne aminokisline. V tem primeru govorimo o ujemanjih. Nekatere aminokisline pa so drugačne med obema proteinoma. Govorimo o neujemanjih. Zato, da lahko optimalno poravnamo obe zaporedji pa moramo celo vnesti vrzeli. Vrzeli so rezultat delecij ali insercij v genu, ki so se zgodile tekom evolucije. Kadar sta si zaporedji zelo podobni, to je takrat kadar je veliko ujemanj, malo pa neujemanj in vrzeli, govorimo da sta proteina homologa in imata skupnega prednika. Hemoglobin in mioglobin sta en tak primer homolognih proteinov. Če še pogledamo zaporedja b-podenot hemoglobina pri drugih vretenčarskih organizmih vidimo, da so si zaporedja njihovih b-podenot prav tako zelo podobna. Takšna primerjava je prikazana na sliki 7 s poravnavo večih zaporedij.
Poravnava je pobarvana na poseben način, kjer so popolnoma ohranjene aminokisline v vseh zaporedjih obarvane s črno barvo, tiste, ki pa so ohranjene v vsaj polovici zaporedij pa s sivo barvo. Razlike med posameznimi zaporedji so nastale tekom evolucije zaradi mutacij v genu potem, ko so se posamezne vrste že ločile med seboj. Takšnim homologom, ki opravljajo isto funkcijo (v našem primeru prenos kisika po telesu) v različnih organizmih, pravimo ortologi. Lahko pa je tekom evolucije prišlo do podvojevanja genov znotraj enega samega organizma potem, ko so se vrste že ločile med seboj. Kasneje so lahko ti podvojeni geni pridobili nove funkcije. Takšne homologe imenujemo paralogi. Primer sta ravno hemoglobin in mioglobin. Hemoglobin sodeluje pri prenašanju kisika po krvožilju, mioglobin pa je v mišičnih vlaknih in tam sprejme kisik od hemoglobina. S konstrukcijo takšnih poravnav večih proteinov lahko nekaj povemo o evolucijskih dogodkih, ki so privedli do današnjega stanja, kako in v kakšnem vrstnem redu so se proteini spreminjali ipd.
Dve zaporedji lahko poravnamo na različne načine. Poravnave poskušamo vedno ovrednotiti, zato da lahko ločimo dobro poravnavo od slabe. Ujemanjem, neujemanjem in vrzelim pripišemo neke vrednosti, iz njihovega števila pa izračunamo rezultat poravnave. Le-ta je za naš primer izpisan na vrhu poravnave na sliki 6 (rezultat poravnave, ang. score). Poravnave so statistično ovrednotene zato, da lahko sklepamo na to ali sta proteina homologa in je podobnost med njima rezultat nekih evolucijskih procesov ali pa sta si proteina podobna zaradi naključnih dogodkov. Pri razvoju algoritmov in programov za poravnave in statistično ovrednotenje poravnav so pomembno vlogo igrali bioinformatiki. V sedemdesetih letih so opisali prve algoritme za primerjave dveh zaporedij. Needleman-Wunsch algoritem je bil prvi, ki je omogočil optimalno poravnavanje dveh zaporedij. Algoritem je omogočil preverjanje vseh možnih poravnav, izbrana pa je bila najbolj optimalna, tista z največjim rezultatom poravnave. V sedemdesetih letih so tudi izračunali kakšne so vrednosti za ujemanja, neujemanja in vrzeli ter te vrednosti zapisali v posebnih tabelah, ki jih imenujemo ocenjevalne matrike. Poznamo kar nekaj ocenjevalnih matrik, ki temeljijo na različnih lastnostih aminokislin in jih lahko pred začetkom iskanja določimo pri nastavitvah iskanja. Ker so bile podatkovne zbirke v sedemdesetih in osemdesetih letih sorazmerno majhne, za preiskavo niso porabili dosti računalniškega časa. Ko se je njihova velikost močno povečala in so konec osemdesetih let po zaslugi interneta postale dostopne tako rekoč vsakomur, so razvili dva nova algoritma, ki sta čas iskanja močno skrajšala. BLAST in FASTA algoritma uporabljamo tudi danes in sta sestavna dela podatkovnih zbirk. BLAST programe lahko najdemo na spletnih straneh NCBI-ja.
Preiskovanje podatkovnih zbirk je danes opravilo, ki mu ne more uiti noben biokemik ali molekularen biolog. Z zbranimi informacijami si lahko zelo pomagamo pri svojem raziskovalnem delu. Preiskovanje podatkovnih zbirk ima vedno večji pomen tudi v farmacevtski industriji, kjer poskušajo poiskati nove proteine, udeležene v različnih bolezenskih stanjih in določiti njihove lastnosti.
Postgenomsko obdobje- prihodnost bioinformatike
Kaj pa sledi v prihodnosti? Kot sem prikazal smo v slabih štiridesetih letih spoznali zakonitosti zgradbe proteinskih molekul in DNA, se naučili določati njihovo zaporedje in pridobili ogromno količino zaporedij. Počasi smo prešli v post-genomsko obdobje, ko smo začeli pojasnjevati vlogo napovedanih genov različnih genomskih projektov. Poleg tega se pozornost znanstvenikov počasi seli na višje nivoje organiziranosti. Zanima nas kako posamezni proteini med seboj sodelujejo, s katerimi proteini se povezujejo, kdaj se izražajo itn. V razvoju je cela vrsta disciplin, ki razvijajo metode za pridobivanje in analizo novih tipov podatkov. Tako se v zadnjih letih govori vedno več o novih znanstvenih disciplinah kot so npr. transkriptomika, proteomika in interaktomika. Tudi za njih velja, da je rast podatkov eksponencialna, da jih združujemo v posebne specializirane podatkovne zbirke in da je omogočeno iskanje in brskanje po zbirkah. Bioinformatiki sodelujejo na vseh stopnjah in pomagajo predvsem z urejanjem podatkov v podatkovnih zbirkah in z računalniško podporo. Nasplošno pa za vse velja, da se v osnovi spreminja način raziskovanja. Bistvo je sedaj v tem, da poskušajo čim hitreje pridobiti čimveč podatkov, šele v naslednji fazi pa iz teh podatkov izluščiti neke zakonitosti (ang. data mining). Torej ne gre več za klasičen način raziskovanja, kjer raziskovalec postavi hipotezo in jo nato poskuša potrditi ali ovreči z eksperimenti. Hipoteze zdaj postavljajo šele v drugi fazi raziskovanja, ko že izluščijo določene vzorce iz nakopičenih podatkov.
Brez dvoma bo poznavanje algoritmov in programov, ki omogočajo obdelavo in iskanje bioloških podatkov v prihodnosti zelo dragoceno, saj lahko močno skrajša čas in s tem posledično tudi ceno raziskav. V zadnjih letih se kažejo potrebe po strokovnjakih s takšnim profilom, predvsem v farmacevtski industriji, ki se v zadnjih letih ukvarja z interpretacijo človeškega genoma, iskanjem genov, ki so udeleženi v boleznih in razvoju zdravil zanje. V tujini že obstajajo študijski programi bioinformatike, kjer načrtno vzgajajo takšne strokovnjake. Tudi pri nas vedno več raziskovalcev pri svojih raziskavah rutinsko preiskuje informacijo shranjeno v podatkovnih zbirkah. Bioinformatika postaja del nepogrešljivih znanj, ki jih mora obvladati raziskovalec v biologiji, ne glede na to ali je biokemik, molekularni biolog, mikrobiolog… Zato smo bioinformatiko začeli poučevati tudi v Sloveniji na Fakulteti za kemijo v okviru dodiplomskega študija Biokemije in na podiplomskem študiju Bioloških znanosti na Biotehnični fakulteti v Ljubljani.
Če se vrnemo na primer z začetka prispevka, bodo v prednosti tisti, ki bodo imeli dobre zemljevide in popolne telefonske imenike, tako da bodo v velikem mestu sposobni hitro poiskati neko informacijo, npr. najboljši servis za domači avto. Tako bodo v postgenomskem obdobju v prednosti tisti raziskovalci in farmacevtska podjetja, ki bodo obvladali orodja za preiskovanje zaporedij proteinov ter nukleinskih kislin in bili sposobni poiskati nove, še neodkrite proteine, vpletene v različne bolezni.
Gregor Anderluh
Slovarček izrazov:
- Bioinformatika – znanstvena disciplina, ki vključuje matematične, statistične in računalniške metode za reševanje bioloških problemov z uporabo nukleotidnih (v DNA) in aminokislinskih (v proteinih) zaporedij in z njimi povezanimi informacijami.
- BLAST, FASTA – posebni algoritmi, ki omogočajo zelo hitro iskanje v primarnih podatkovnih zbirkah s proteinskimi ali nukleotidnimi zaporedji.
- Genomski projekti – projekti, s katerimi želijo določiti nukleotidno zaporedje celotnega genoma, t.j. celotne dednine, posebej izbranih organizmov. Prvi določen evkariontski genom je bil genom kvasovke S. cerevisiae. Prvi izsledki projekta človeški genom so bili objavljeni leta 2001. Zaradi količine podatkov so to izredno zahtevni projekti, ki jih izvajajo družno posebej specializirani centri po vsem svetu.
- Homologni proteini – proteini, ki imajo skupnega prednika. Tekom evolucije je prihajalo do mutacij v genih in proteini se zato lahko zelo razlikujejo v primarni zgradbi. če sta si dva proteina iz različnih organizmov zelo podobna (imata ohranjenih veliko aminokislin), potem pravimo, da sta homologa. Homologni proteini so lahko ortologi (opravljajo isto funkcijo v dveh različnih organizmih) ali paralogi (so nastali s podvojitvijo genov po speciaciji in lahko pridobijo nove funkcije).
- Interaktomika – znanstvena disciplina, ki raziskuje omrežja povezav proteinov in genov v živih celicah.
- Poravnavanje zaporedij – primerjava dveh ali več zaporedij DNA ali proteinov. V zaporedja lahko vnesemo vrzeli, tako da lahko poravnamo ekvivalentne položaje na isto mesto. Poravnave so osnovna metoda analize zaporedij in jih uporabljamo predvsem za iskanje najbolj ohranjenih delov zaporedij, ki imajo lahko nek biološki pomen.
- Primarna zgradba proteina – zaporedje aminokislin, ki gradijo protein. Aminokisline zapišemo s posebno enočrkovno kodo, kjer vsaka črka predstavlja svojo aminokislino.
- Proteomika – znanstvena disciplina, ki se ukvarja z izraženim komplementom genoma (proteini). Poskuša razumeti, kateri proteini so izraženi v določenem obdobju v različnih tipih celic. Raziskovalci v proteomiki uporabljajo predvsem dvo-dimenzionalne poliakrilamidne gele, na katerih ločujejo vse proteine v celicah naenkrat na podlagi njihovega naboja in velikosti.
- Sekundarne podatkovne zbirke – podatkovne zbirke, ki vsebujejo informacije, pridobljene iz zaporedij proteinov ali DNA. Največkrat so v obliki profilov, motivov ali blokov. To so kratki koščki zaporedij, ki so enaki ali najbolj podobni (ohranjeni) deli poravnave več zaporedij. Uporabljamo jih predvsem pri opisu proteinskih družin.
- Tridimenzionalna zgradba proteina – določen položaj vseh aminokislin proteina v prostoru.
- Transkriptomika – znanstvena disciplina, ki se ukvarja z izraženim komplementom genoma (mRNA). Za razliko od proteomike transriptomika deluje na ravni mRNA. Raziskovalci uporabljajo predvsem izražene dele nukleotidnih zaporedij (ang. EST clones) ali genske čipe za celosten opis aktivnosti celice.
Literatura:
- Attwood TK, Parry-Smith DJ. Introduction to Bioinformatics. Harlow, United Kingdom: Prentice Hall; 1999.
- Baxevanis AD, Francis Ouellette BF. Bioinformatics A Practical Guide to the Analysis of Genes and Proteins. New York, USA: Wiley-Interscience, A John Wiley & Sons Inc., Publication; 1998.
- Higgins D, Taylor W. Bioinformatics: Sequence, Structure, and Databanks. Oxford, United Kingdom: Oxford University Press; 2000.
- Kanehisa M. Post-genome Informatics. Oxford, United Kingdom: Oxford University Press; 2002.
- Bioinformatika v biologiji (Predavanja za podiplomski študij bioloških in biotehnoloških znanosti na Biotehniški fakulteti.)
Slike:
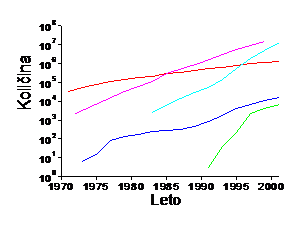
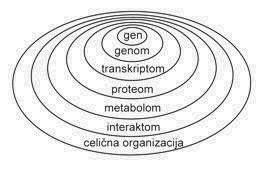
Slika 1: Razvoj bioloških in računalniških znanostih lahko ponazorimo s strmino krivulje, ki opisuje število dogodkov v časovnem obdobju (leva stran slike). Razvoj v računalništvu je prikazan s povečevanjem števila tranzistorjev na čipu, ki se podvoji vsako leto in pol (vijolična krivulja; t.i. Moore-ov zakon, vir). Razvoj interneta je prikazan na slovenskem primeru kot povečevanje števila domen registriranih pri Arnesu (zelena krivulja, vir Arnes). Hitro kopičenje bioloških podatkov je prikazano z rastjo števila nukleotidnih zaporedij v podatkovni zbirki GenBank (svetlo modra krivulja, vir) in s povečevanjem števila znanih 3D struktur proteinov ali nukleinskih kislin v podatkovni zbirki Protein Data Bank (temno modra krivulja, vir ). Prikazana je tudi rast člankov, ki so označeni s ključno besedo Biokemija v bibliografski podatkovni zbirki PubMed (rdeča krivulja, vir). Sočasen razvoj obeh panog se kaže s podobnim naklonom krivulj.
Desno so prikazani različni nivoji organiziranosti živega znotraj katerih delujejo znanstveniki v genomskem in post-genomskem obdobju. Večina bioloških podatkov je trenutno na prvih dveh osnovnih nivojih, nivoju genov in genomov. Prirejeno po Vihinen Mauro (Bioinformatics in proteomics, Biomolecular Engineering 18 (2001) 241-248).
|
|
|

|

|
|

|

















{kind=link}